What are we going to learn?
During this session, you will:
- analysis of variance (ANOVA) in base R
- linear models in base R
- the tidymodel approach
Essential shortcuts
Remember some of the most commonly used RStudio shortcuts:
- function or dataset help: press F1 with your cursor anywhere in a function name.
- execute from script: Ctrl + Enter
- assignment operator (
<-): Alt + -
Material
Load the following packages:
library(tidymodels) # for parsnip package and rest of tidymodels
# helper packages
library(readr) # for importing data
library(car) # Companion to Applied Regression package
library(performance) # Assessment of Regressmon Models performance
library(dotwhisker)# for visualizing regression results
library(parsnip)Remember to use Ctrl+Enter to execute a command from the script.
About the data
The following section will be using data from Constable (1993) to explore how three different feeding regimes affect the size of sea urchins over time.
Sea urchins reportedly regulate their size according to the level of food available to regulate maintenance requirements. The paper examines whether a reduction in suture width (i.e., connection points between plates; see Fig 1 from constable 1993) is the basis for shrinking due to low food conditions.

Figure 1 from Constable 1993 paper showing sea urchin plates and suture width
The data in csv format is available from the tidymodels website and were assembled for a tutorial here.
urchins <-
# read in the data
read_csv("https://tidymodels.org/start/models/urchins.csv") %>%
# change the names to be more description
setNames(c("food_regime", "initial_volume", "width")) %>%
# convert food_regime from chr to a factor, helpful for modeling
mutate(food_regime = factor(food_regime,
levels = c("Initial", "Low", "High")))urchins # see the data as a tibble## # A tibble: 72 x 3
## food_regime initial_volume width
## <fct> <dbl> <dbl>
## 1 Initial 3.5 0.01
## 2 Initial 5 0.02
## 3 Initial 8 0.061
## 4 Initial 10 0.051
## 5 Initial 13 0.041
## 6 Initial 13 0.061
## 7 Initial 15 0.041
## 8 Initial 15 0.071
## 9 Initial 16 0.092
## 10 Initial 17 0.051
## # ... with 62 more rowsWe have 72 urchins with data on:
- experimental feeding regime group with 3 levels (Initial, Low, or High)
- size in milliliters at the start of the experiment (initial_volume)
- suture width in millimeters at the end of the experiment (width, see Fig 1)
{kind=link}
Statistics in R using base and stats
Visualize the data
Use a boxplot to visualize width versus food_regime as a factor and a scatterplot for width versus initial_volume as a continuous variable.
boxplot(width ~ food_regime, data = urchins)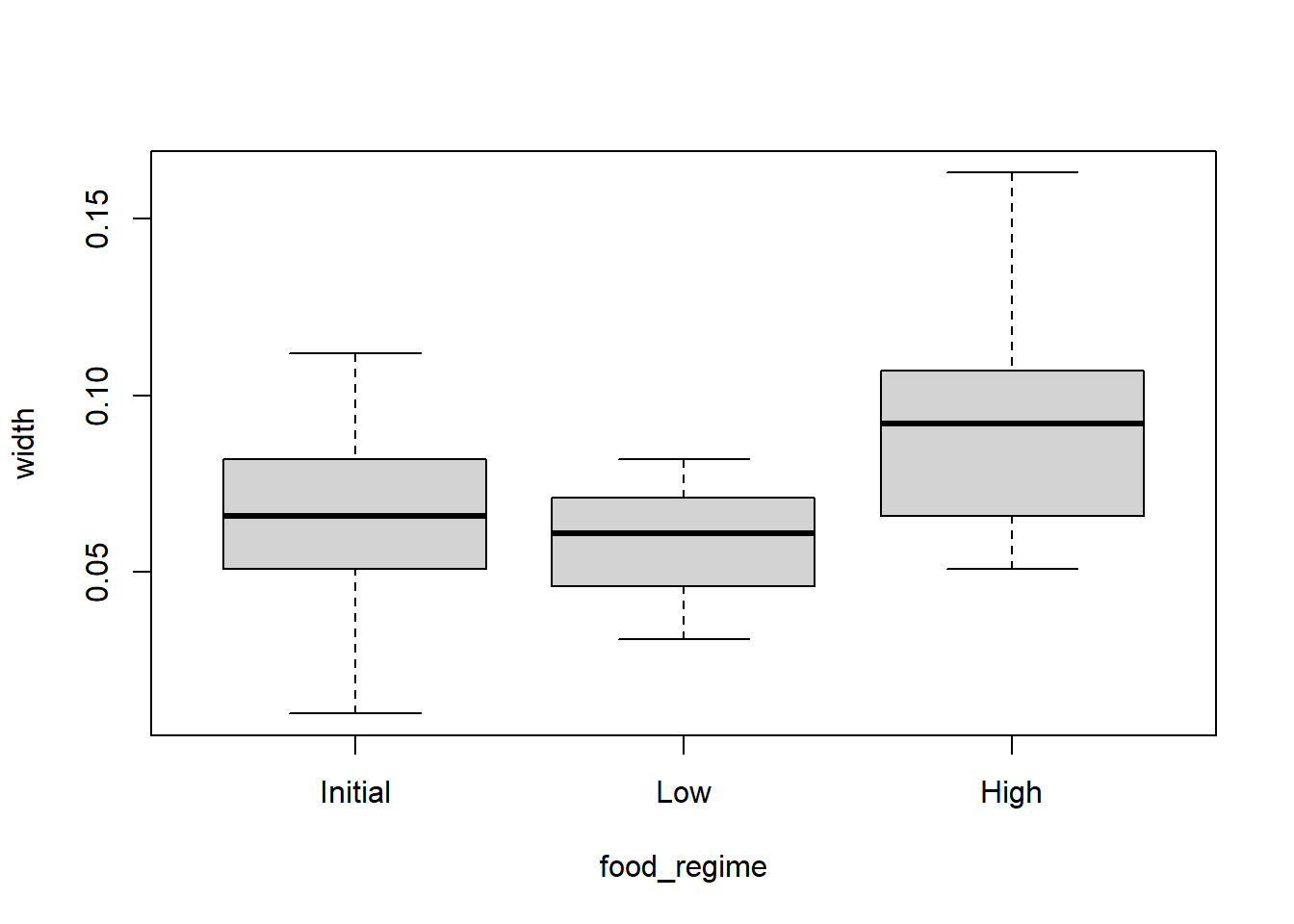
plot(width ~ initial_volume, data = urchins)
We can see that there are some relationships between the response variable (width) and our two covariates (food_regime and initial volume). But what about the interaction between the two covariates?
Challenge 1 - use ggplot2 to make a plot visualizing the interaction between our two variables. Add a trendline to the data.
Hint: think about grouping and coloring.
ggplot(urchins,
aes(x = initial_volume,
y = width,
group = food_regime,
col = food_regime)) +
geom_point() +
geom_smooth(method = lm, se = FALSE) +
scale_colour_viridis_d(option = "plasma", end = 0.7)## `geom_smooth()` using formula 'y ~ x'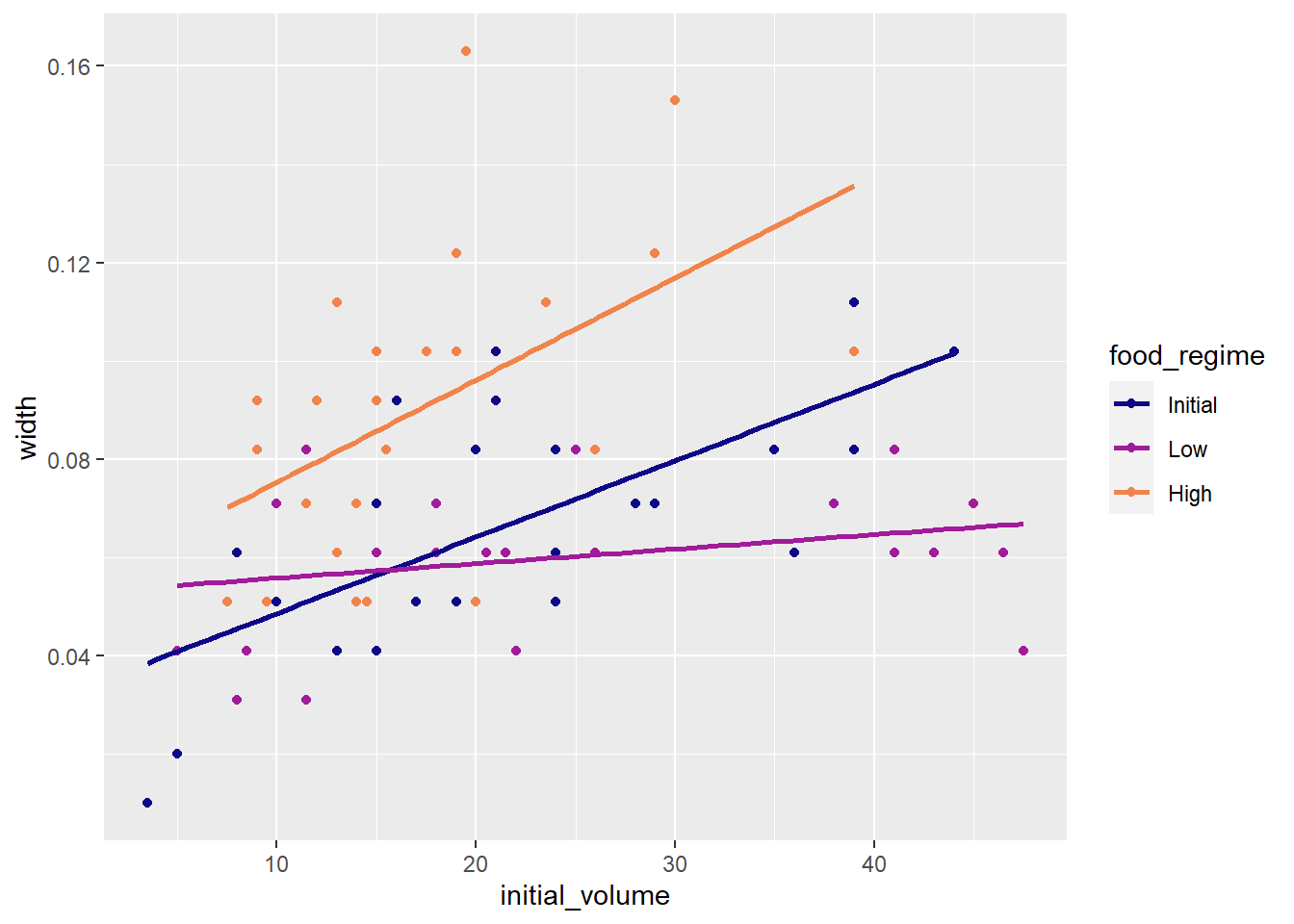
Urchins that were larger in volume at the start of the experiment tended to have wider sutures at the end. Slopes of the lines look different so this effect may depend on the feeding regime indicating we should include an interaction term.
Analysis of Variance (ANOVA)
Information in this section was taken from rpubs.com and Data Analysis in R Ch 7.
We can do an ANOVA with the aov() function to test for differences in sea urchin suture width between our groups. We are technically running and analysis of covariance (ANCOVA) as we have both a continuous and a categorical variable. ANOVAs are for categorical variables and we will see that some of the post-hoc tests are not amenable to continuous variables.
aov()uses the model formularesponse variable ~ covariate1 + covariate2. The * denotes the inclusion of both main effects and interactions which we have done below. The formula below is equivalent toreponse ~ covar1 + covar2 + covar1:covar2i.e. the main effect of covar 1 and covar 2, and the interaction between the two.
aov_urch <- aov(width ~ food_regime * initial_volume,
data = urchins)
summary(aov_urch) # print the summary statistics## Df Sum Sq Mean Sq F value Pr(>F)
## food_regime 2 0.012380 0.006190 13.832 9.62e-06 ***
## initial_volume 1 0.008396 0.008396 18.762 5.15e-05 ***
## food_regime:initial_volume 2 0.004609 0.002304 5.149 0.00835 **
## Residuals 66 0.029536 0.000448
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Both the main effects and interaction are significant (p < 0.05) indicating a significant interactive effect between food regime and initial volume on urchin suture width. We need to do a pairwise-comparison to find out which factor levels and combination of the two covariates have the largest effect on width.
Pair-wise comparison
Run a Tukey’s Honestly Significant Difference (HSD) test - note it does not work for non-factors as per the warning message.
TukeyHSD(aov_urch)## Warning in replications(paste("~", xx), data = mf): non-factors ignored:
## initial_volume## Warning in replications(paste("~", xx), data = mf): non-factors ignored:
## food_regime, initial_volume## Warning in TukeyHSD.aov(aov_urch): 'which' specified some non-factors which will
## be dropped## Tukey multiple comparisons of means
## 95% family-wise confidence level
##
## Fit: aov(formula = width ~ food_regime * initial_volume, data = urchins)
##
## $food_regime
## diff lwr upr p adj
## Low-Initial -0.006791667 -0.021433881 0.007850548 0.5100502
## High-Initial 0.023791667 0.009149452 0.038433881 0.0006687
## High-Low 0.030583333 0.015941119 0.045225548 0.0000129The comparison between High-Initial and High-Low food regimes are significant (p < 0.05).
Checking the model
We also want to check that our model is a good fit and does not violate any ANOVA assumptions:
- Data are independent and normally distributed.
- The residuals from the data are normally distributed.
- The variances of the sampled populations are equal.
Challenge 2 - Use a histogram and qqplots to visually check data are normal.
hist(urchins$width)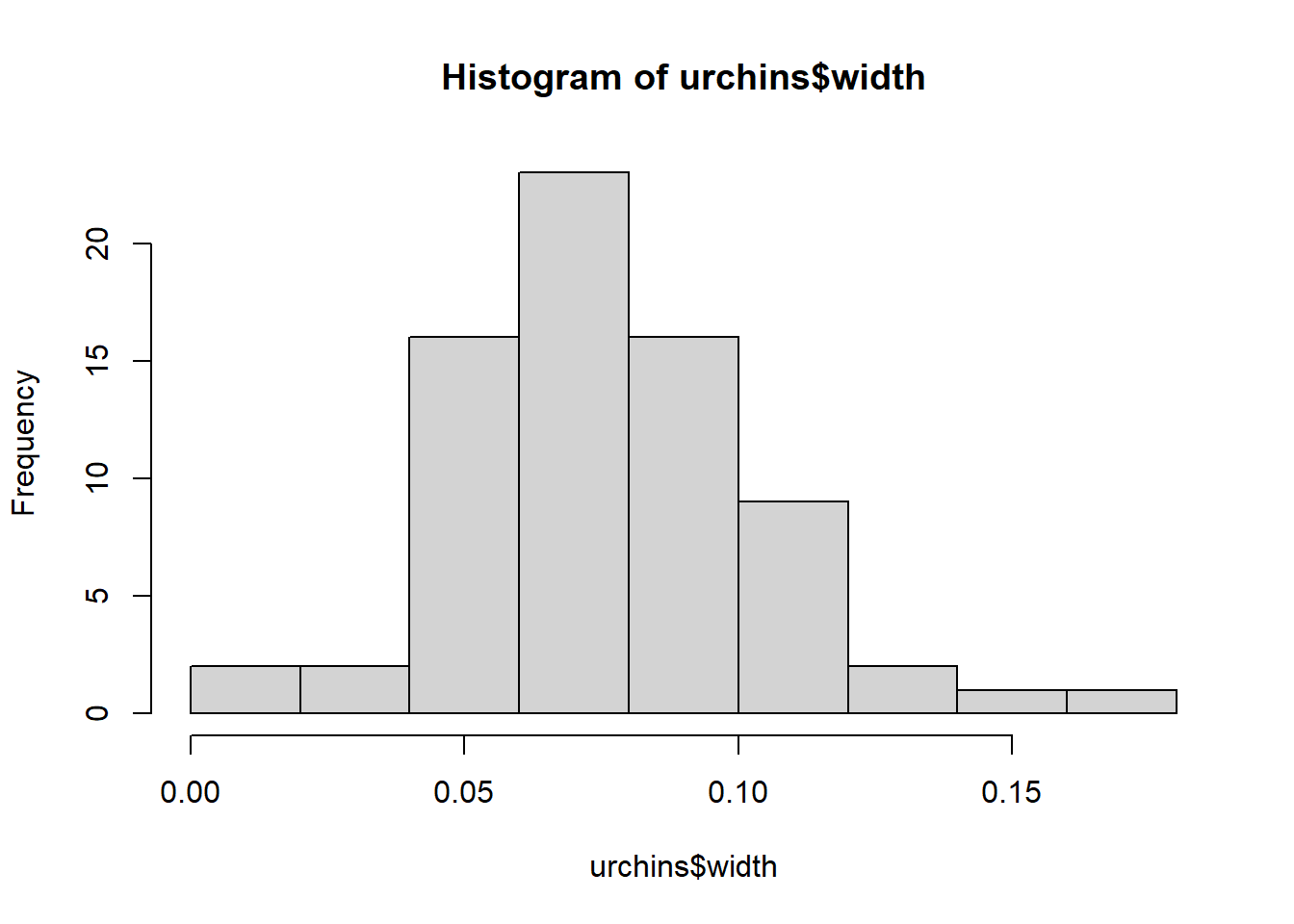
qqnorm(urchins$width)
qqline(urchins$width)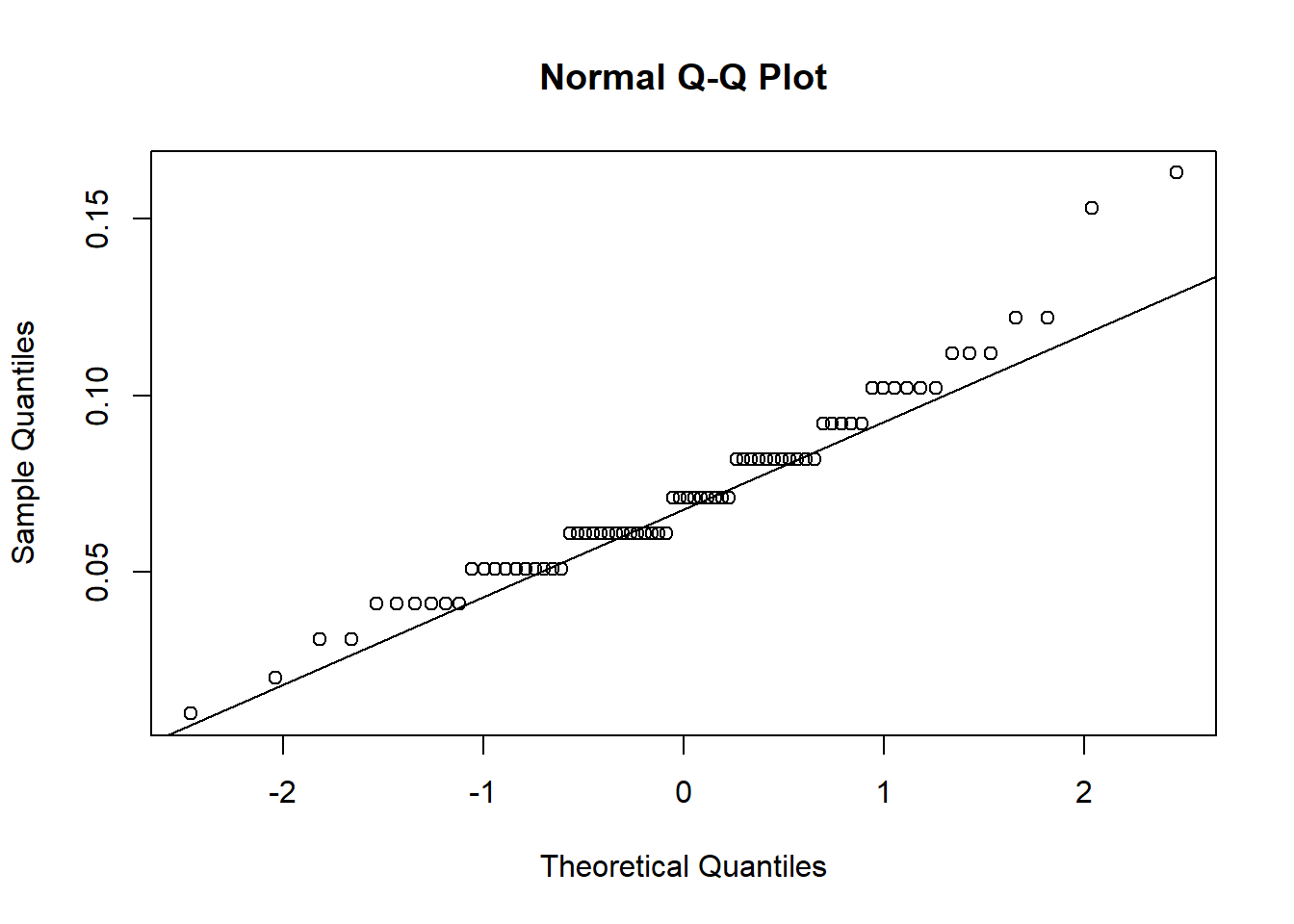
You could also run a Shapiro-Wilk test on the data:
shapiro.test(urchins$width)##
## Shapiro-Wilk normality test
##
## data: urchins$width
## W = 0.95726, p-value = 0.01552Check the model residuals. Plot the residuals vs fitted values - do not want too much deviation from 0.
plot(aov_urch, 1)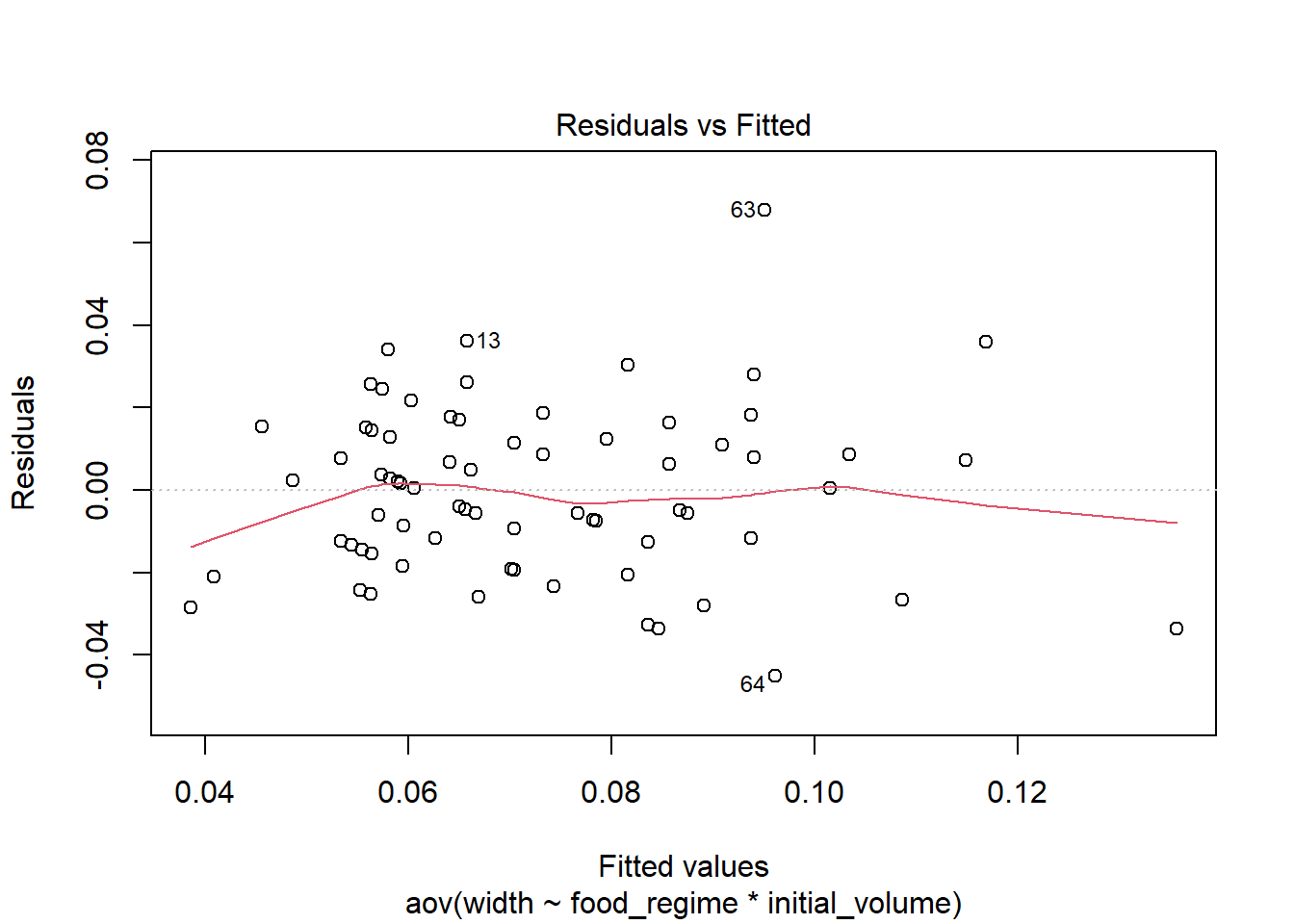
plot(predict(aov_urch) ~ aov_urch$fitted.values)
abline(0, 1, col = "red")
Check the normality of residuals, run Shapiro-Wilk test on residuals:
plot(aov_urch, 2)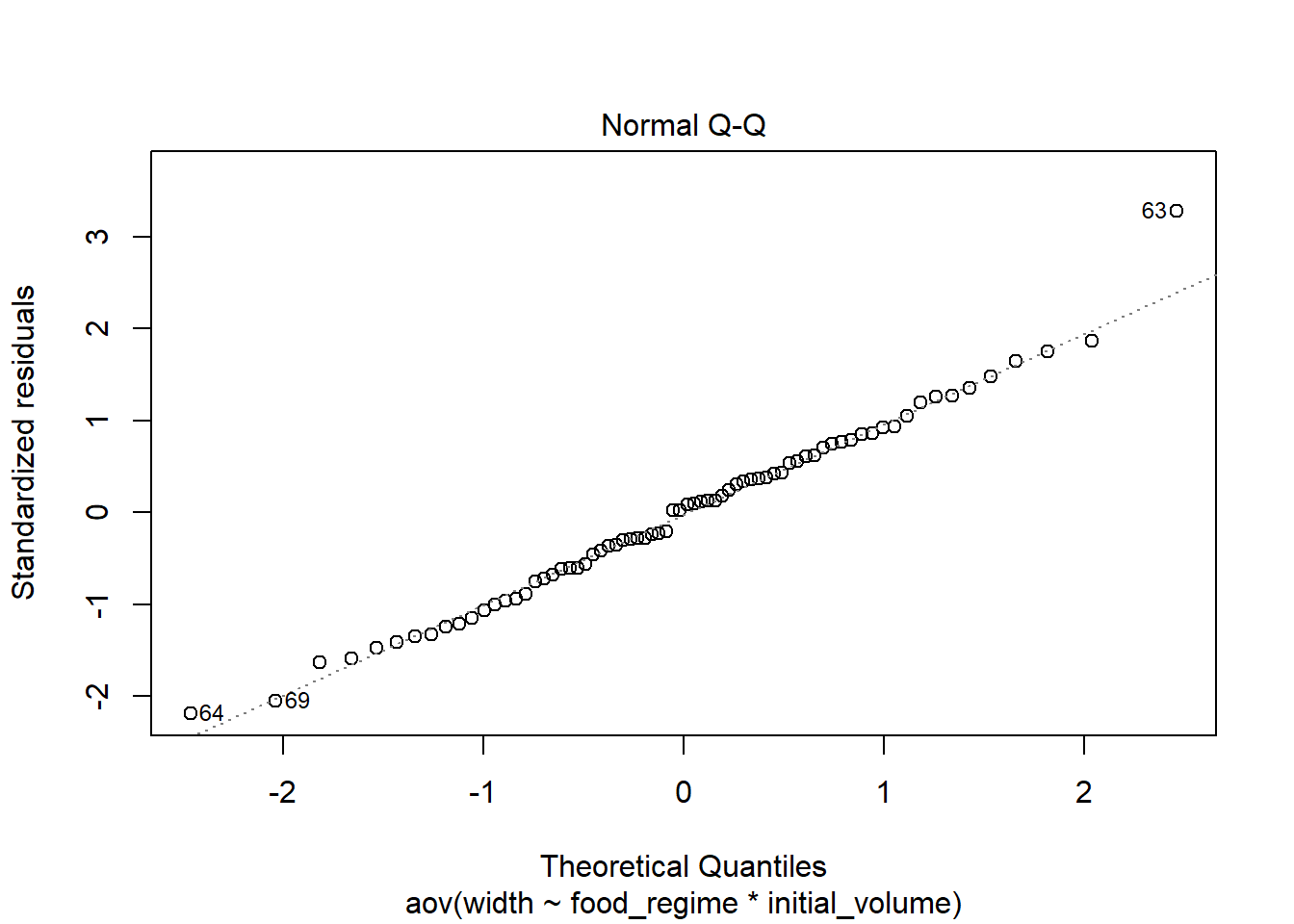
shapiro.test(resid(aov_urch))##
## Shapiro-Wilk normality test
##
## data: resid(aov_urch)
## W = 0.98456, p-value = 0.5244The residuals fall on the Normal Q-Q plot diagonal and the Shapiro-Wilk result is non-significant (p > 0.05).
Check for homogeneity of variance
Challenge 3 - use the help documentation for leveneTest() from the car package to check homogenetity of variance on food_regime.
Again, only works for factor groups.
leveneTest(width ~ food_regime, data = urchins)## Levene's Test for Homogeneity of Variance (center = median)
## Df F value Pr(>F)
## group 2 4.4224 0.01559 *
## 69
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1The Levene’s Test is significant for food_regime (not what we want) and there are a few options to deal with this. You can ignore this violation based on your own a priori knowledge of the distribution of the population being samples, drop the p-value significance, or use a different test.
Linear Model
lm_urch <- lm(width ~ food_regime * initial_volume,
data = urchins)
summary(lm_urch)##
## Call:
## lm(formula = width ~ food_regime * initial_volume, data = urchins)
##
## Residuals:
## Min 1Q Median 3Q Max
## -0.045133 -0.013639 0.001111 0.013226 0.067907
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 0.0331216 0.0096186 3.443 0.001002 **
## food_regimeLow 0.0197824 0.0129883 1.523 0.132514
## food_regimeHigh 0.0214111 0.0145318 1.473 0.145397
## initial_volume 0.0015546 0.0003978 3.908 0.000222 ***
## food_regimeLow:initial_volume -0.0012594 0.0005102 -2.469 0.016164 *
## food_regimeHigh:initial_volume 0.0005254 0.0007020 0.748 0.456836
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.02115 on 66 degrees of freedom
## Multiple R-squared: 0.4622, Adjusted R-squared: 0.4215
## F-statistic: 11.35 on 5 and 66 DF, p-value: 6.424e-08In the output, we have the model call, residuals, and the coefficients. The first coefficient is the (Intercept) and you might notice the food_regimeInitial is missing. The function defaults to an effects parameterization where the intercept is the reference or baseline of the categorical group - Initial in this case.
>You can change the reference level of a factor using the relevel() function.
The esimates of the remaining group levels of food_regime represents the effect of being in that group. To calculate the group coefficients for all group levels you add the estimates for the level to the intercept (first group level) estimate. For example, the estimate for the ‘Initial’ feeding regime is 0.0331 and we add the estimate of ‘Low’ (0.0331 + 0.0197) to get the mean maximum size of 0.0528 mm for width.
For the continuous covariate, the estimate represents the change in the response variable for a unit increase in the covariate. ‘Initial Volume’s’ estimate of 0.0015 represents a 0.0015 mm increase (the estimate is positive) in width per ml increase in urchin initial volume.
We can get ANOVA test statistics on our linear model using the anova() in base or Anova() from the car package.
anova(lm_urch)## Analysis of Variance Table
##
## Response: width
## Df Sum Sq Mean Sq F value Pr(>F)
## food_regime 2 0.0123801 0.0061900 13.8321 9.616e-06 ***
## initial_volume 1 0.0083962 0.0083962 18.7621 5.154e-05 ***
## food_regime:initial_volume 2 0.0046088 0.0023044 5.1494 0.008354 **
## Residuals 66 0.0295358 0.0004475
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Anova(lm_urch)## Anova Table (Type II tests)
##
## Response: width
## Sum Sq Df F value Pr(>F)
## food_regime 0.0168653 2 18.8434 3.358e-07 ***
## initial_volume 0.0083962 1 18.7621 5.154e-05 ***
## food_regime:initial_volume 0.0046088 2 5.1494 0.008354 **
## Residuals 0.0295358 66
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1These are effectively the same as the aov() model we ran before.
Note: The statistics outputs are the same comparing the
aov()andanova()models while theAnova()model is not exactly the same. TheAnova()output tells us it was a Type II test and theaov()documentation says it is only for balanced designs which means the Type 1 test is the applied (see here). The type of test can be set forAnova()but not the others. Here, the overall take-away from the different ANOVA functions are comparable.
Challenge 4 - use the check_model() documentation to apply the function to our lm_urch model.
The performance package has a handy function check_model() that will check several aspects of your model in one go:
check_model(lm_urch)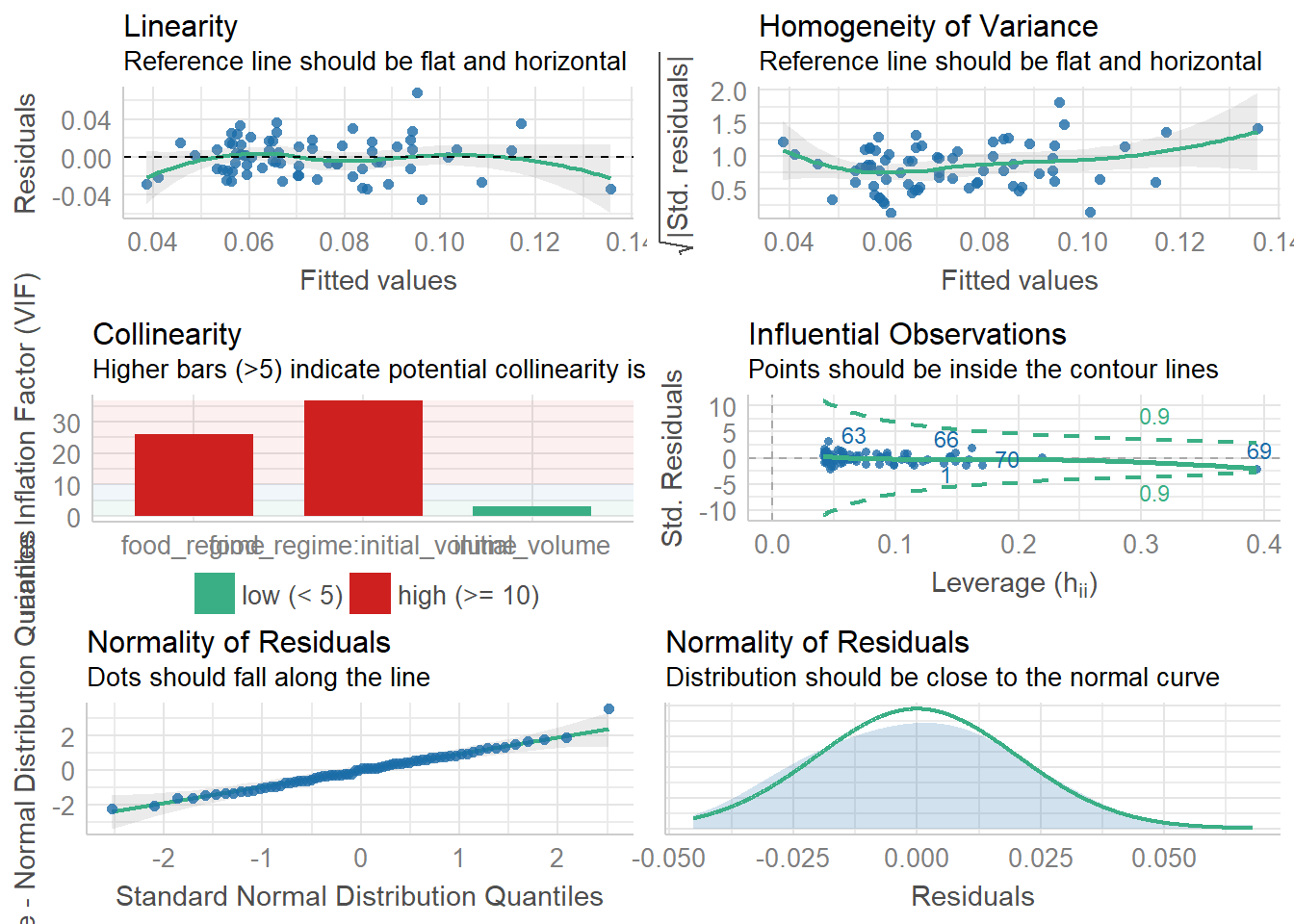
Introducing Tidymodels
Like the tidyverse package, the tidymodels framework is a collection of packages for modeling and machine learning following the tidyverse principles.
This section is modified from the first Tidymodels article.
Build and fit a model
Let’s apply a standard two-way analysis of variance (ANOVA) model to the dataset as we did before. For this kind of model, ordinary least squares is a good initial approach.
For Tidymodels, we need to specify the following:
- The functional form using the parsnip package.
- The method for fitting the model by setting the engine.
We will specify the functional form or model type as “linear regression” as there is a numeric outcome with a linear slope and intercept. We can do this with:
linear_reg() ## Linear Regression Model Specification (regression)
##
## Computational engine: lmOn its own, not that interesting. Next, we specify the method for fitting or training the model using the set_engine() function. The engine value is often a mash-up of the software that can be used to fit or train the model as well as the estimation method. For example, to use ordinary least squares, we can set the engine to be lm.
The documentation page for linear_reg() lists the possible engines. We’ll save this model object as lm_mod.
lm_mod <-
linear_reg() %>%
set_engine("lm")Next, the model can be estimated or trained using the fit() function and the model formula we used for the ANOVAs:
width ~ initial_volume * food_regime
lm_fit <-
lm_mod %>%
fit(width ~ initial_volume * food_regime, data = urchins)
lm_fit## parsnip model object
##
## Fit time: 0ms
##
## Call:
## stats::lm(formula = width ~ initial_volume * food_regime, data = data)
##
## Coefficients:
## (Intercept) initial_volume
## 0.0331216 0.0015546
## food_regimeLow food_regimeHigh
## 0.0197824 0.0214111
## initial_volume:food_regimeLow initial_volume:food_regimeHigh
## -0.0012594 0.0005254We can use the tidy() function for our lm object to output model parameter estimates and their statistical properties. Similar to summary() but the results are more predictable and useful format.
tidy(lm_fit)## # A tibble: 6 x 5
## term estimate std.error statistic p.value
## <chr> <dbl> <dbl> <dbl> <dbl>
## 1 (Intercept) 0.0331 0.00962 3.44 0.00100
## 2 initial_volume 0.00155 0.000398 3.91 0.000222
## 3 food_regimeLow 0.0198 0.0130 1.52 0.133
## 4 food_regimeHigh 0.0214 0.0145 1.47 0.145
## 5 initial_volume:food_regimeLow -0.00126 0.000510 -2.47 0.0162
## 6 initial_volume:food_regimeHigh 0.000525 0.000702 0.748 0.457This output can be used to generate a dot-and-whisker plot of our regression results using the dotwhisker package:
tidy(lm_fit) %>%
dwplot(dot_args = list(size = 2, color = "black"),
whisker_args = list(color = "black"),
vline = geom_vline(xintercept = 0,
color = "grey50",
linetype = 2))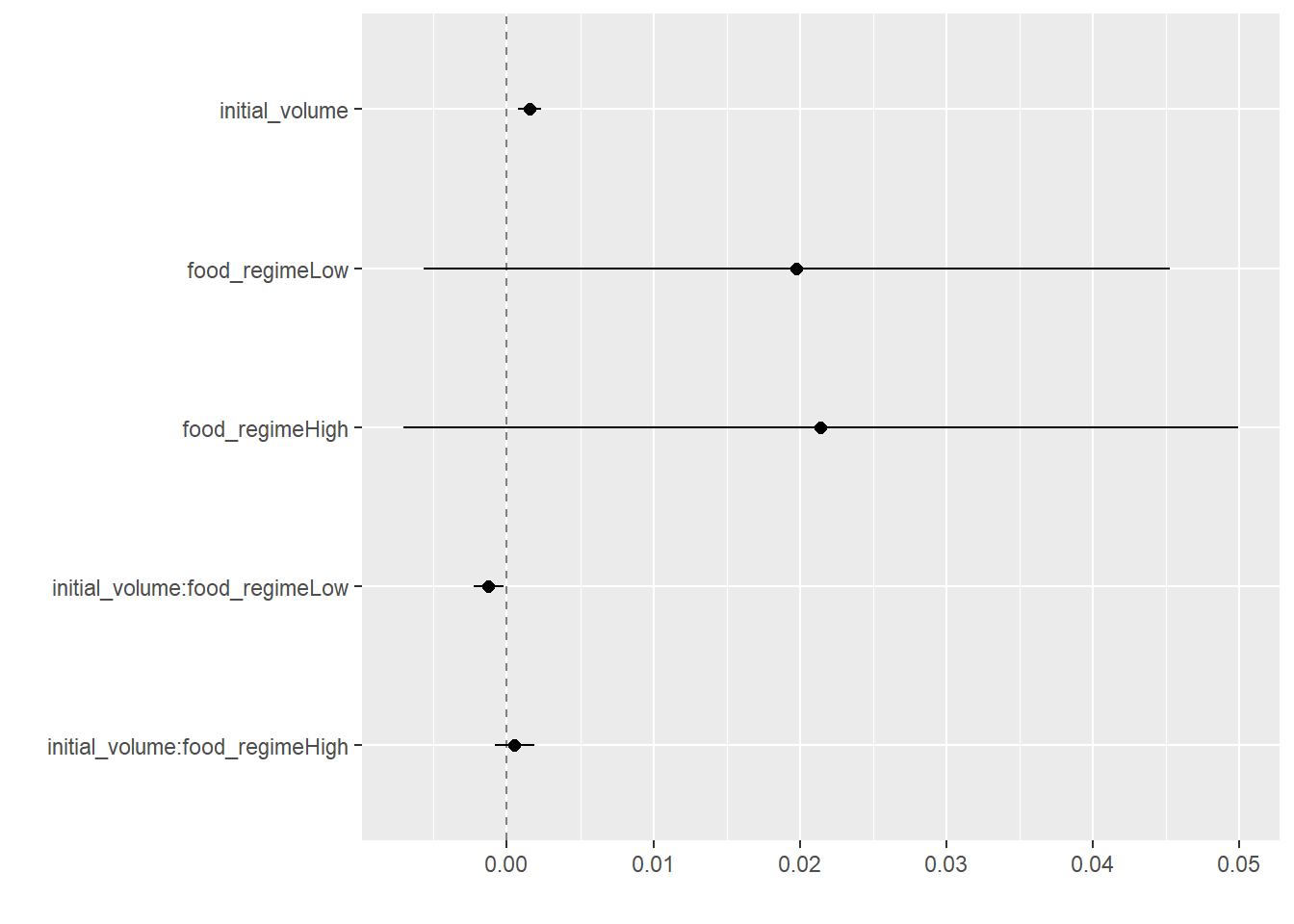
Use a model to predict
Say that it would be interesting to make a plot of the mean body size for urchins that started the experiement with an initial volume of 20 ml.
First, lets make some new example data to predict for our graph:
new_points <- expand.grid(initial_volume = 20,
food_regime = c("Initial", "Low", "High"))
new_points## initial_volume food_regime
## 1 20 Initial
## 2 20 Low
## 3 20 HighWe can then use the predict() function to find the mean values at 20 ml initial volume.
With tidymodels, the types of predicted values are standardized so that we can use the same syntax to get these values.
Let’s generate the mean suture width values:
mean_pred <- predict(lm_fit, new_data = new_points)
mean_pred## # A tibble: 3 x 1
## .pred
## <dbl>
## 1 0.0642
## 2 0.0588
## 3 0.0961When making predictions, the tidymodels convention is to always produce a tibble of results with standardized column names. This makes it easy to combine the original data and the predictions in a usable format:
conf_int_pred <- predict(lm_fit,
new_data = new_points,
type = "conf_int")
conf_int_pred## # A tibble: 3 x 2
## .pred_lower .pred_upper
## <dbl> <dbl>
## 1 0.0555 0.0729
## 2 0.0499 0.0678
## 3 0.0870 0.105# now combine:
plot_data <-
new_points %>%
bind_cols(mean_pred, conf_int_pred)
plot_data## initial_volume food_regime .pred .pred_lower .pred_upper
## 1 20 Initial 0.06421443 0.05549934 0.07292952
## 2 20 Low 0.05880940 0.04986251 0.06775629
## 3 20 High 0.09613343 0.08696233 0.10530453# and plot:
ggplot(plot_data,
aes(x = food_regime)) +
geom_point(aes(y = .pred)) +
geom_errorbar(aes(ymin = .pred_lower,
ymax = .pred_upper),
width = .2) +
labs(y = "urchin size")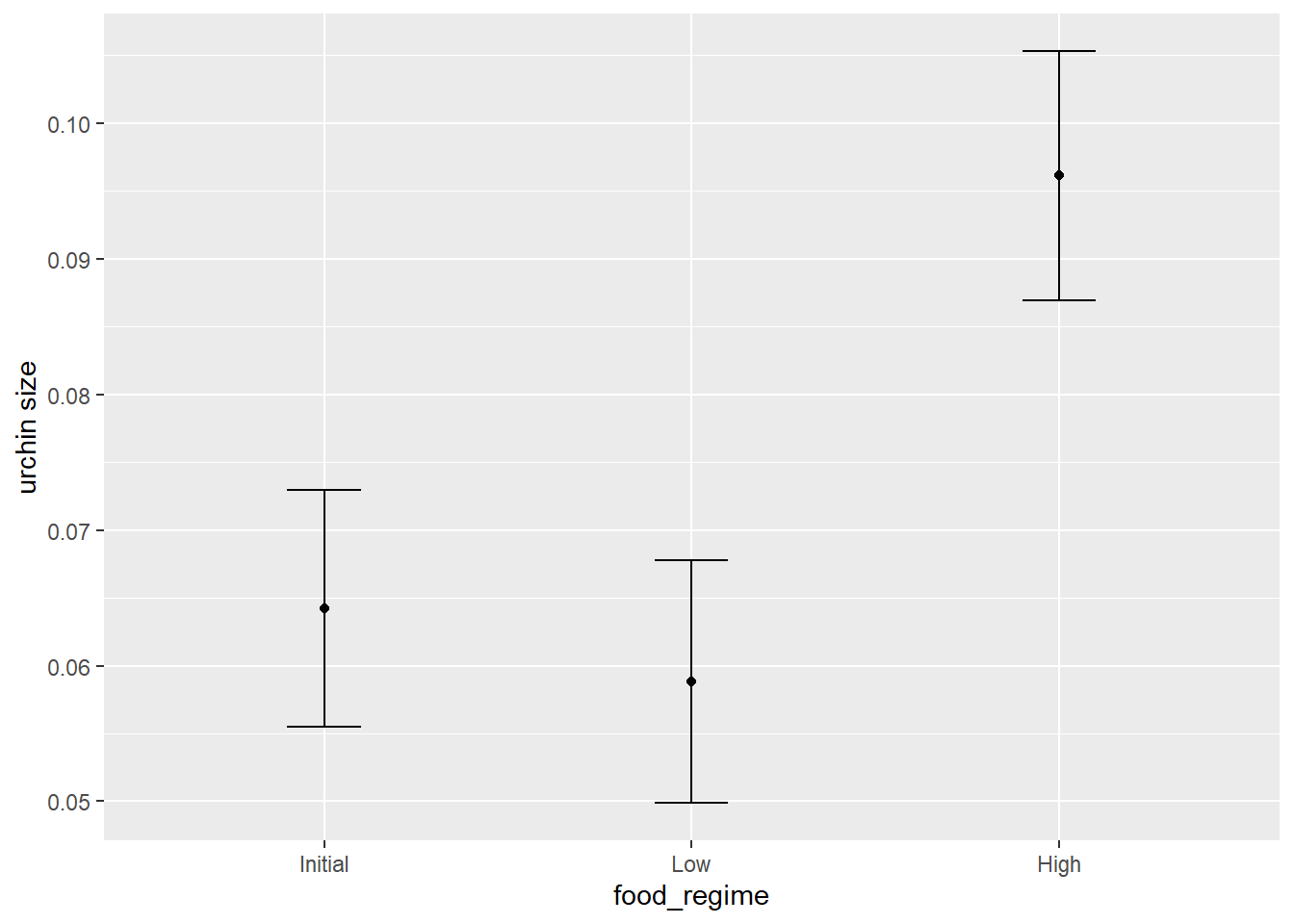
There is also an example of a Bayesian model in the tidymodels article I have not included here.
Close project
Closing RStudio will ask you if you want to save your workspace and scripts. Saving your workspace is usually not recommended if you have all the necessary commands in your script.
Useful links
- For statistical analysis in R:
- Steve Midway’s Data Analysis in R Part II Analysis
- Jeffrey A. Walker’s Applied Statistics for Experiemental Biology
- Chester Ismay and Albert Y. Kim’s ModernDive Statistical Inference via Data Science
- For tidymodels:
- Our compilation of general R resources